
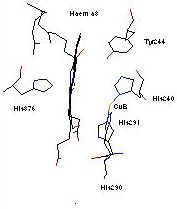
Heme-copper oxidases, including cytochrome c oxidase (CcO), catalyze the four-electron, four-proton reduction of dioxygen to water, while also ‘pumping’ four protons (per O2 molecule reduced) across the cell membrane. The electrochemical potential gradient generated by this process ultimately provides the driving force for ATP synthesis. The key chemistry for O2-binding, reduction, and coupled proton translocation occurs at the binuclear heme a3- CuB active-site. Recent protein x-ray structures show the copper ion ligated to three imidazole (from histidine) N-donor groups, with the high spin heme a3 being bound to an imidazole from histidine which is distal to copper (Figure).
The goals of our model studies for heme-copper oxidases are to develop chemistry relevant to (i) the O2-binding and reduction chemistry, (ii) the effects of protonation and/or addition of electrons (as in the enzyme), and (iii) the numerous studies carried out on oxidized resting state of the enzyme, which may give Fe-X-Cu species (X = oxide, hydroxide, cyanide, azide, carboxylate).
In our initial attempts to delve into this type of chemistry, we chose to use TMPA-copper complexes, with known O2-chemistry (see above). When (3) and (1a) react with O2 (1/2 equivalent), [(F8-TPP)FeIII-(O2-)-CuII(TMPA)]+ (4) is isolated. This chemistry already represents a crude functional model for CcO, since O2 has been reduced to the oxidation state level of water; isotope labeling studies indicate the bridging oxo group in (4) comes from O2. Interestingly, the acid-base reaction of equimolar amounts of (5), (6), and Et3N gives the same complex, (4).
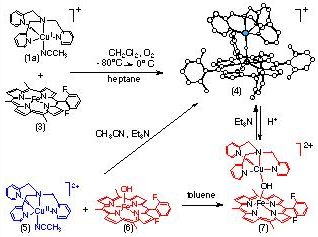
The x-ray structure of (7) reveals a linear Fe(III)-oxo-Cu(II) coordination (angle of Fe-O-Cu = 178.2?, with very short Fe-O and Cu-O bond lengths. The complex has also been characterized by UV-Vis, IR, Mssbauer, and NMR spectroscopies, as well as magnetic susceptibility measurements. The 1H-NMR spectrum is very rich, with resonances ranging from +65 ppm downfield to -104 ppm upfield. A detailed NMR study has been published (ref. # 146) and the unusual behavior is ascribed to the S = 2 ground state with strong antiferromagnetic coupling between Fe(III) and Cu(II) ions.
Protonation of (4) by addition of one equivalent of triflic acid produces the hydroxo-bridged complex, [(F8-TPP)Fe-(OH)-Cu(TMPA)]2+ (7). Structural insights from x-ray absorption spectroscopy reveal elongated Fe-OH and Cu-OH bonds and a bending such that angle of Fe-O-Cu at 157? Complex (7) is a plausible model for the resting (‘as-isolated) oxidized enzyme, with its supposed Fe-X-Cu antiferromagnetic coupling. Since protonation of enzyme turnover intermediates leads to product water and/or translocated protons, it is of interest that the oxo moiety in (4) is basic; Protonation experiments suggest that pKa ~ 8 ?2.5 in water.
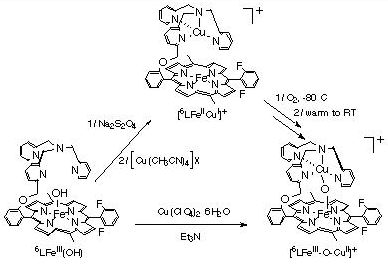
While the dioxygen chemistry and mechanism of formation of (4) from (1a) and (3) is of interest, we have chosen to emphasize related studies on binucleating ligands, where a tetradentate or tridentate donor for copper (or another metal ion) is synthetically tethered to the periphery of a porphyrin. Recent work with the 6L (see Figure) indicates analogous chemistry affords a di-iron species with 6L (ref. 136) and [(6L)FeIII-OH], with empty tether. Reaction of [(6L)FeIII-OH] with a Cu(II) salt in presence of a base results in the formation of the new oxo-bridged complex [(6L)FeIII-O-CuII]+, which has been characterized by UV-Vis and 1H-NMR spectroscopies, mass spectrometry, and X-ray diffraction (see below). Preliminary observations show that a reduced species [(6L)FeII-CuI]+ can be generated, and that its subsequent reaction with O2 leads to [(6L)FeIII-O-CuII]+. [(6L)FeIII-O-CuI]+ is prepared by reduction of [(6L)FeIII-OH] with dithionite followed by addition of a copper(I) salt. It is characterized by lambda(max) = 424 nm, and by 1H-NMR properties consistent with high-spin Fe(II) in tetrahydrofuran (THF) as solvent. Interestingly, when the oxygenation of this reduced compound is carried out at -80 �C in THF, a new spectroscopically distinct species is observed; this cleanly converts to [(6L)FeIII-O-CuII]+ after warming to room-temperature. These observations bode well for future investigations where we wish to probe the possibility of forming Fe-peroxo-Cu or other interesting intermediates which may have relevance to the processes of O2-binding, reduction, O-O cleavage and proton translocation by the Fe-Cu center in cytochrome c oxidases.
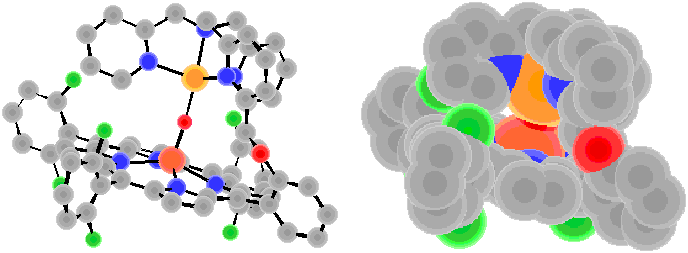
Cu-O =1.851 ?Fe-O =1.746 ?
Fe…Cu = 3.586 ?
angle of Fe-O-Cu = 170.9?br>
Thus, ongoing projects in the heme-copper oxidase model chemistry area are as follows:
1) We wish to investigate new dioxygen-metal chemistry, wherein (porphyrinate)-iron(II) with (ligand)-copper(I) complexes react with O2. Do Fe-(O2)-Cu complexes form? If so, what are their structures and how does this depend on (i) the particular porphyrin, (ii) copper ligation (e.g., tri or tetradentate ligands), and (iii) the presence or absence of a distal axial base (ligand) for the heme-iron.
2) What is the effect of protonation or addition of electrons (i.e., reduction) on such Fe-(O2)-Cu species. Can hydro(peroxo) metal and/or ferryl (FeIV=O) (or perhaps FeV=O) intermediates be detected and studied. Such intermediates have been seen or proposed for the enzyme O2-reduction chemistry. Our model systems and chemistry at low-temperatures should be ideal for such studies.
3) What is the effect of having phenol-ligands for copper. Recent enzyme x-ray structural updates suggest there is a covalent bond between His-240 and a nearby Tyr-244 (see Figure, above) around CuB. The functional implications of this for the O2-reduction and/or proton pumping mechanism have yet to be deduced, but one possibility suggested is that this His-Tyr ‘dimer’ could donate an electron to dioxygen, becoming a phenoxyl radical. Thus the CuB site effectively donates two electrons, one from Cu(I) and one from the phenol. We are currently designing ligand systems and reactivity studies to investigate this kind of new chemistry.
4) We are also pursuing the chemistry of heme/non-heme di-iron complexes. We find that initial metallation (with Fe) of our binucleating ligands like 6L (or an analogue isomer 5L) can lead to complexes such as [(5L)FeIII-O-FeIII-Cl]+ (not shown; see publication # 136). Such compounds and their chemistry may represent good models for the active site of Nitric oxide (NO) Reductase, which converts NO to N2O; this enzyme is evolutionarily related to heme-copper oxidases. We are currently generated reduced heme/non-heme diiron(II) compounds for such reactivity studies; in terms of getting biomimetic reactivity, initial results look very promising.
5) A series of synthetic heterobinucleating ligands are being synthesized and studied, possessing a tridentate (rather than tetradentate) ligand tethered to a porphyrin periphery. These also have an axial base, a pyridine donor, covalently attached. From previous studies, we anticipate tridentate vs tetradentate Cu chelation will dramatically and interestingly alter the chemistry obseved, while the ‘internal’ axial base will model the enzyme proximal histidine Fe ligation.