
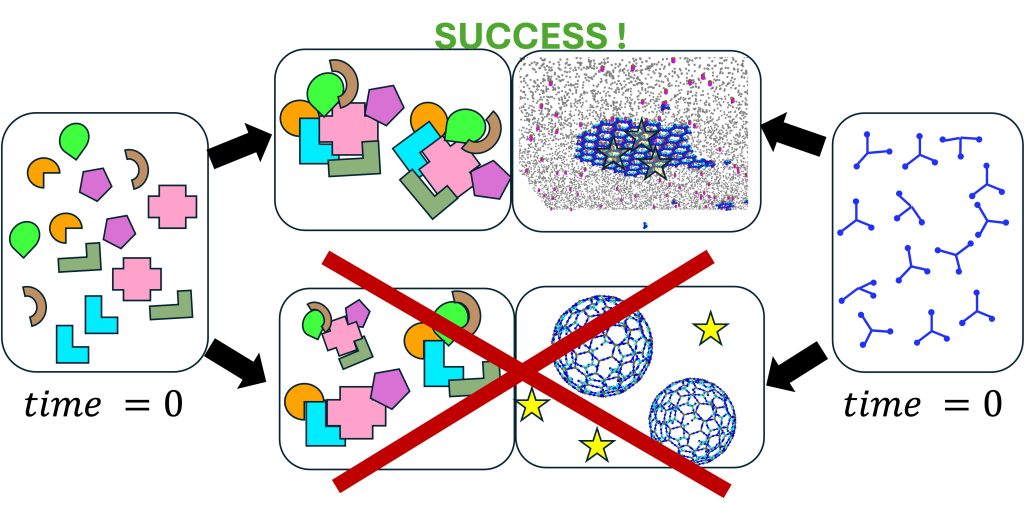
Self-assembly and self-organization in cell biology
Recent papers:
Discovering optimal kinetic pathways for self-assembly using automatic differentiation, Jhaveri et al, PNAS 2024.
Membrane-associated self-assembly for cellular decision making, preprint, Foley et al, 2025
A membrane-driven biochemical oscillator tunable by the volume to surface area ratio, preprint Fischer et al, 2025
In our research group we study dynamical systems in biology, both in and out of equilibrium. We address questions related to control of self-assembly in living systems, which has to occur at the right time and place for proper function. We are especially fascinated by clathrin-mediated endocytosis, viral exit from cells, and DNA transcription and maintenance. Our multidisciplinary group tackles questions both on principles and design of self-assembling systems, as well as biological function in cells. To study these complex systems, we simulate networks of many interacting components, and construct simplified models that are amenable to analytical theory, with foundations in statistical mechanics. We use and develop modeling techniques from physics, chemistry, and engineering, including molecular models for thermodynamics, reaction-diffusion models for stochastic dynamics in cells, continuum models for coupling to mechanics, and ML for optimization. We collaborate frequently with experimental groups.
Spatial and temporal control of vesicle formation in clathrin-mediated endocytosis
Recent work:
Membrane-associated self-assembly for cellular decision making, preprint, Foley et al, 2025
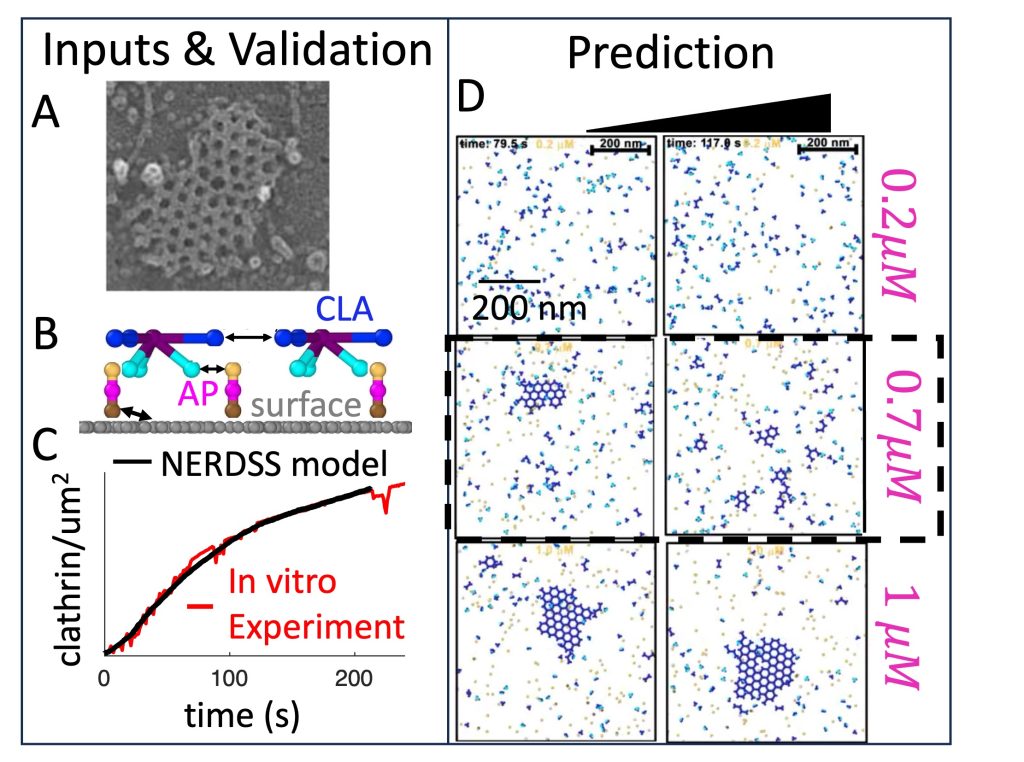
Large self-assembled clathrin lattices spontaneously disassemble without sufficient adaptor proteins, Guo et al, PLos Comp Biol, 2022.
Dynamin isoforms exploit distinct recruitment patterns to endocytic clusters, Jiang et al, Nat Comm 2024.
Clathrin-mediated endocytosis is occurring persistently within our cells, performing transport of essential nutrients such as iron into our cells, and clearing receptors at our neural synapse. By controlling the receptor composition at the membrane surface, endocytosis dynamically responds to signals, impacting cell state and motility. It involves coordinated assembly of dozens of different protein components, and viral infection or neurological disorders can result from its dysfunction. Our research has shown how localization to the membrane can dramatically enhance assembly of even very weakly interacting partners via dimensionality reduction from 3D to 2D, which effectively concentrates components and promotes binding. At the systems level, we have quantified how the stoichiometry of the network of protein and lipid components involved in CME can sensitively tune the speed or success of vesicle formation, highlighting again the central role of membrane localization in triggering protein assembly. With reaction-diffusion simulations, we have quantified how essential lipid phosphatases are capable of driving disassembly of the membrane-bound assemblies. Ongoing research in our lab is focused on predicting how the membrane composition and environment controls the frequency and success of vesicle formation for productive receptor uptake and healthy cell function. NIH MIRA
Viral assembly, exit, and membrane remodeling
Recent Work:
Temporal control by co-factors prevents kinetic trapping, Qian et al, Biophys J, 2023
Modeling membrane reshaping driven by dynamic protein assemblies, Fu et al, Curr Opin Struc Biol, 2023
Membrane bending energy selects for symmetric growth of protein assemblies, Ying, Y. et al, Submitted, 2025.
Predicting protein curvature sorting across membrane compositions, Fu et al, Biophys. J 2026
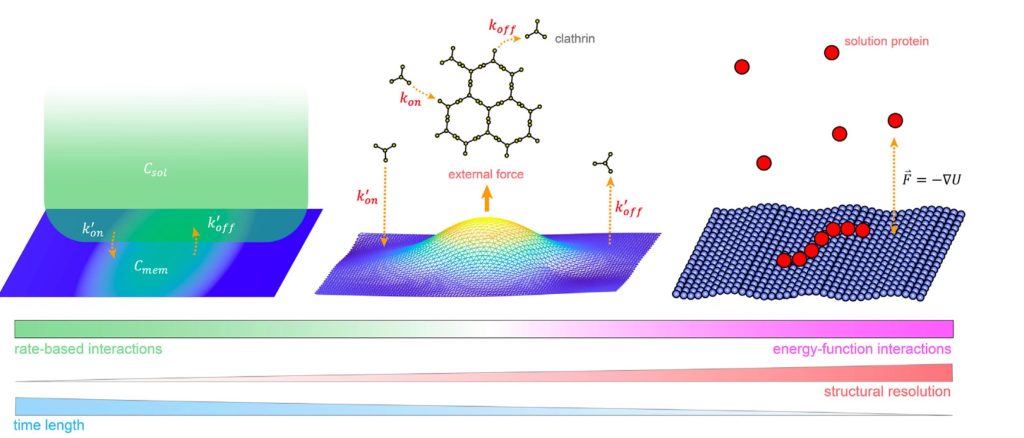
For entry in and exit from the cell, remodeling of the membrane is almost universally driven by self-assembling proteins. To understand the mechanics and dynamics of this remodeling as it couples to the largely biochemically driven interactions between proteins, we develop continuum membrane models and apply them to a range of processes in cell biology (Fu, 2021) (Fu, 2023). A prime example is viral exit, where in HIV infection, the retroviral Gag protein must assemble at the plasma membrane to bud out and form new infectious virions. We have studied several key steps in the life-cycle of the HIV virus, including how co-factors control the Gag assembly within cells (Qian, 2023), and how timing of remodeling of the Gag lattice in budded virions is tuned for maturation (Guo, 2023). We are integrating our membrane models with our self-assembly software to model the coupled dynamics of self-assembly and budding. NSF CAREER, NIH MIRA, NIAID R01.
Software for modeling of non-equilibrium self-assembly
Recent Work:
Parallelization of particle-based reaction-diffusion simulations using MPI. Guo et al, J Comput Chem, 2025.
ioNERDSS: Transforming macromolecular structures into simulations of self-assembly preprint and web server, 2026
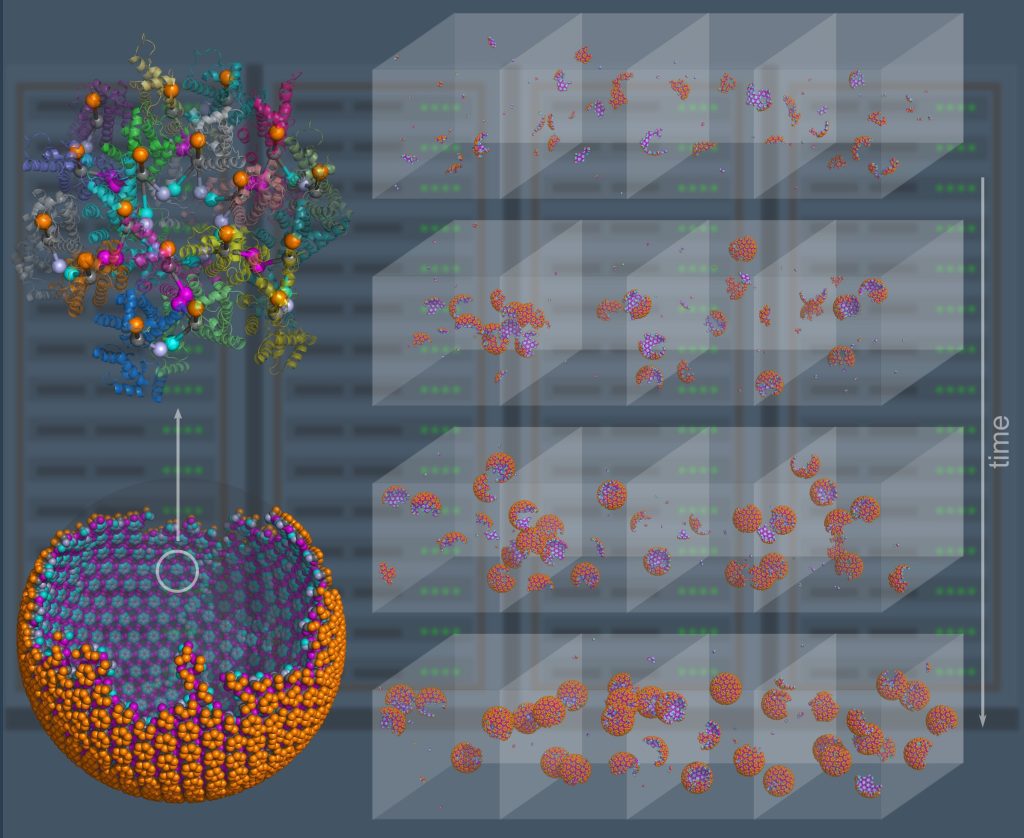
NERDSS: a nonequilibrium simulator for multibody self-assembly at the cellular scale, Varga et al, Biophys J, 2020.
Computer simulation provides a powerful complement to experiment because we can track the spatial and temporal behavior of individual proteins at nm and sub-μs resolution, respectively, and produce a model of the collective protein dynamics. There are a number of challenges in trying to model these processes with both the efficiency to reach long (min) time-scales and the resolution to capture the structural features of a growing protein complex. We developed novel single-particle and rigid-body reaction-diffusion (RD) algorithms that combine both efficiency and accuracy in solving the governing equations of motion in 3D, in 2D, and transitioning from 3D to 2D (Yogurtcu et al, 2015) (Fu, 2019). These algorithms have been combined into user-friendly software (Varga, 2020) that has been applied to diverse biological systems like viral formation, clathrin-mediated endocytosis, and silk-fiber formation, with direct comparison to experiment. NIH MIRA. We recently parallelized the code using MPI (Guo, 2025).
Check out the NERDSS web server for Python tools to help set-up and run NERDSS simulations directly from a PDB structure, with our new ioNERDSS python package.
Dimensional reduction in biology
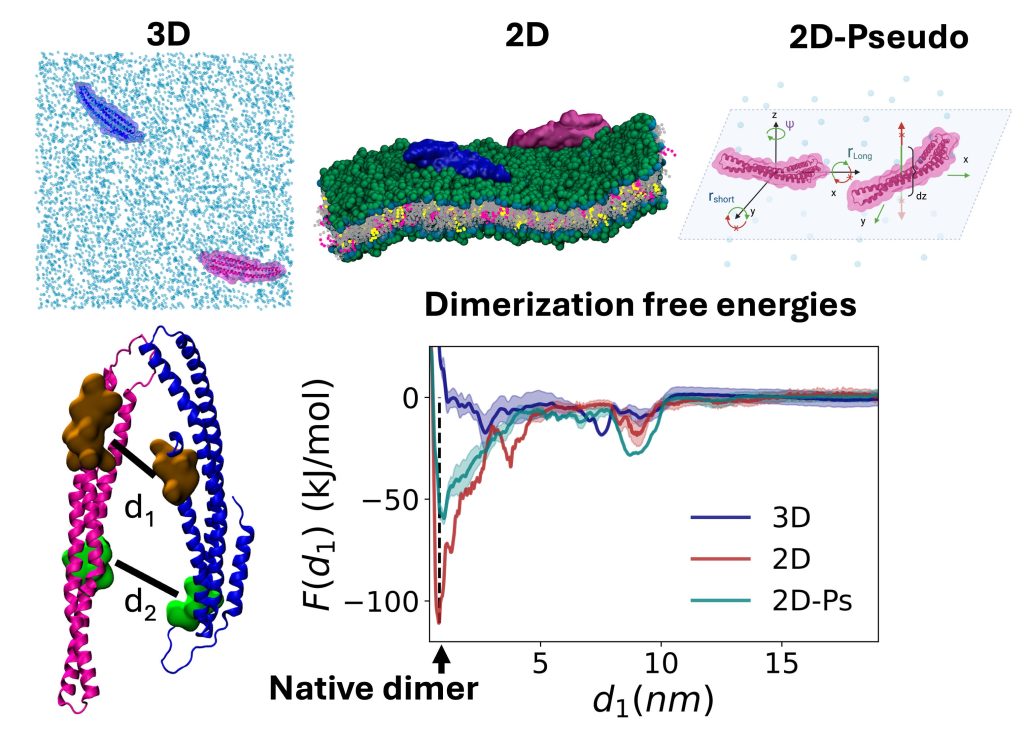
Recent Work:
Protein dimerization in 2D vs 3D: geometric allostery enhances binding affinity, Jhaveri et al, J Chem Phys 2025
Mechanisms of enhanced or impaired DNA target selectivity driven by protein dimerization, Sang et al, PNAS Nexus 2026
Speed limits of protein assembly with reversible membrane localization, Mishra et al, J Chem Phys, 2021.
Proteins frequently localize to lower-dimensional environments in the cell to perform their target function, whether the effectively 2D surface of the membrane, or the 1D road of DNA. This concept of dimensional reduction was established in the 1960s as an effective means for a protein to accelerate its target search (e.g. membrane receptor), by searching in a reduced space that concentrates reactants. We have developed model systems and theory to further quantify how self-assembly, not just target searches, can dramatically benefit from dimensional reduction. We have shown that for physiologically relevant regimes of protein concentrations, binding affinities, cell V/A ratios, and lipid populations, dimensional reduction drives dramatic increases in dimer formation, thereby regulating the initiation of robust assembly (Yogurtcu 2018). Modulating the ‘stickiness’ of the membrane via lipid populations (e.g., from enzymes) will then tune when and where assembly occurs (Varga, 2020). Somewhat surprisingly, we showed the kinetics of dimer formation can also increase by orders-of-magnitude due to 2D localization, even though diffusion on the 2D surface is ~100-fold slower than in solution (Mishra 2021). These same ideas apply to self-assembly along the effectively 1D DNA (Sang 2025). Finally, the dissociation constant in 2D/1D is distinct from 3D, and we characterized using MD simulations that it can be much more favorable due to allosteric changes on the membrane (Jhaveri et al, 2025). NIH MIRA, NIH U01.
Model design from experimental data
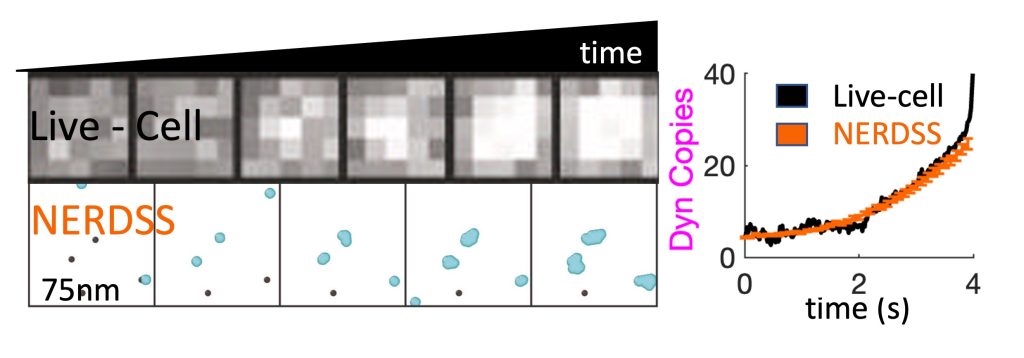
Recent Work:
Dynamin1 long and short-tail isoforms exploit distinct recruitment to endocytic nanoclusters, Jiang et al Nat Comm 2024.
GAGA Zinc finger transcription factor searches chromatin by 1D-3D facilitated diffusion, Feng et al, Nat Struc Mol Biol, 2025
Secretion-Catalyzed Assembly of Protein Biomaterials on a Bacterial Membrane Surface, Xie et al, Angewante Chem, 2023.
A challenge with any cell-scale biological model is defining the requisite reaction network and corresponding rate constants that can quantitatively reproduce in vivo dynamics (Johnson, 2021). We are adopting optimization methods from the machine learning community to enable searches in high-dimensional parameter spaces, similar to what is required in training a neural network (Jhaveri, 2024). These simulations of complex systems also stimulate our development of simpler models amenable to analytical theory. We combined theory and kinetic models of 3-7 subunit complexes to quantify how control of assembly kinetics via optimal selection of rate constants can ensure robust and efficient assembly for multi-subunit complexes that would otherwise suffer from kinetic trapping (Jhaveri, 2024). We seek to improve our model description as constrained by experimental data, such as single-particle tracking data at super-resolution, which effectively monitors the same dynamics as our models, albeit typically just one molecule type at a time. We developed and optimized a model of dynamin clustering and assembly against live-cell sptPALM data, predicting how specific isoforms of dynamin could produce distinct densities of dynamin clusters in response to Calcium stimulation (Jiang, 2024). We are working with our colleagues at Johns Hopkins to apply our methods to quantify experimental data on self-assembly in diverse systems, including DNA transcription (Feng, 2025) and silk-fiber assembly (Xie, 2023). NIH MIRA, NIH U01.