
ASMD
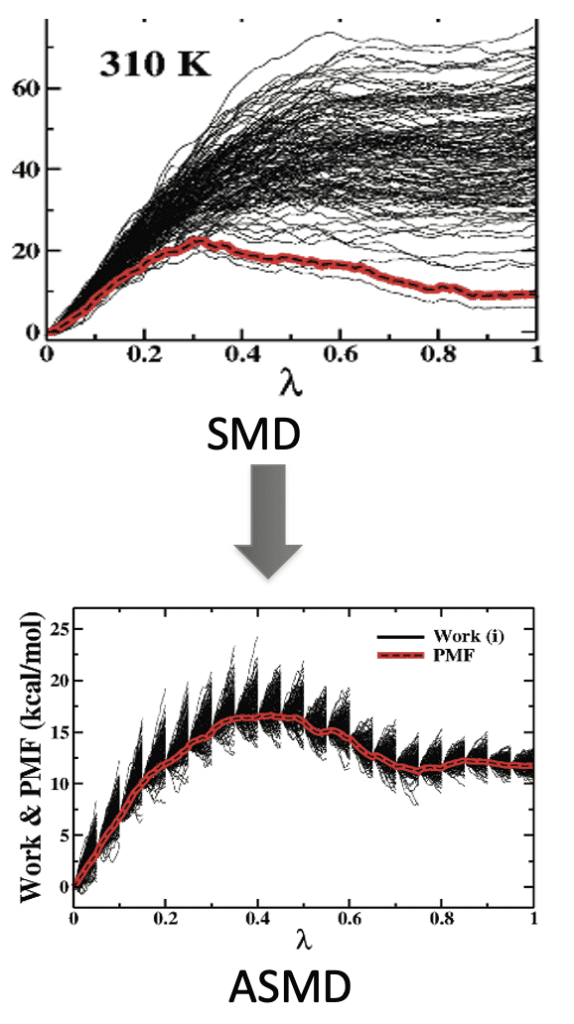
Adaptive Steered Molecular Dynamics (ASMD) is an enhanced sampling method of Steered Molecular Dynamic (SMD) simulations. In SMD, one end of the protein/polymer is fixed, while the other end is attached to a pseudo-atom with a harmonic potential. Then, a force is applied on the pseudo atom to stretch the peptide along a certain coordinate. Due to the need of Jarzynski’s equality calculation, a large number of measurements is needed to get the converged PMF results. ASMD overcomes this concern by dividing the whole reaction coordinate into several small stages to limit the spread of work. In such a way, ASMD reduces the number of trajectories that must be sampled by discarding the trajectories that have deviated far from the equilibrium path in stages. Depending on the different contraction criteria at the end of stages, there are several ASMD variants (e.g., Naive-ASMD, MB-ASMD, FR-ASMD, etc.)Recently, ASMD has been applied successfully to investigate the unfolding process of alanine-rich polypeptides, Neuropeptide Y (NPY) and the relevant mutants.
ASMD has been extended to include a dynamic water box which further increases the efficiency of utilization of super computing resources. We call this as the ASMD with telescoping water box method. In this method the water box is big enough to incorporate the protein in each stage. After the stage is finished the protein is removed and placed in a new water box which is longer to incorporate the amount of protein that would be unraveled in the next stage. The protein is equilibrated in this new water box before being sent to the next stage.
ACM/ACAMM
The ACM/ACAMM research project is about using simulation tools to demonstrate that engineered nanoparticle (ENP) networks can potentially be used as autonomous computing materials in data storage and computing.The overarching goal is trying to capture the structural and dynamic features of ENP assemblies as an analog to biological neuronal network in our brain, based upon the molecular interactions among ENPs. Different from conventional neuromorphic engineering where neuronal algorithms are mapped to silicon-based architect, we are trying to simulate and develop novel material with intrinsic characteristics as neuronal systems.
We are using molecular dynamics (MD) simulations to study the mechanism of polymers interacting nanoparticles, as well as mean-field theory (MFT), coarse-grained MD and Monte Carlo (MC) simulations to study the network connections between ENPs. Our research also involves using MD simulations, classical Poisson-Boltzmann equation and DLVO theory to calculate the electrical potential and zeta-potential of ENPs. We are also working on simulating a signal transport in ENP networks, trying to find out the degree to which the spatio-temporal signals transport through ENP networks and whether they follow the same rules as seen in time-series neuronal signals. The last part of the project involves mathematical modeling of a network, using Competitive Threshold Linear Network (CTLN) models. We work on it as a top-down approach to determine desirable characteristics of a network in terms of its topology and properties, to guide the bottom-up approach work.

Sustainable Nanotechnology
Projects under the sustainable nanotechnology are performed in collaboration with Center for Sustainable Nanotechnology (CSN) (https://susnano.wisc.edu/) Nanomaterials exhibit unique chemical and physical properties due to their small sizes, shapes, and surface coatings. Nanoparticles play a vital role in numerous applications such as medical diagnostics, fuel cells and in energy applications like in solar cells. We partner with CSN to collectively contribute to the goal of understanding molecular-level nanomaterial-biological interactions to develop sustainable and beneficial nanotechnologies. Our objective is to provide computational and theoretical insights and to answer questions related to understanding the nanomaterial properties and nanomaterial-biological interactions using molecular dynamics simulations and machine learning models.
Machine learning offers the potential to explore correlations in experiment or simulation that are not otherwise apparent. Specifically, we hypothesize that it can be used to resolve the connection between underlying molecular scale properties of nanoparticles and their interactions with biomolecules and structures in bacteria to macroscopic functions in the cell, e.g. toxicity and viability. Our group has created a small database from the CSN-published results on nanoparticle-induced bacterial toxicity. Results strongly suggest that artificial neural networks (ANNs) can successfully predict such toxicity. Currently, we confirmed the accuracy and precision of the prediction of nanoparticle viabilities using the optimized bag of ANNs for new data examples and we try to establish the degree to which these predictions are robust to given examples in the data set. We are also working on predicting the optical properties of carbon materials using machine learning techniques.
We use all atom simulations and develop Dissipative Particle Dynamics (DPD) to model proteins (eg. Cytochrome C) with various ligand-wrapped gold nanoparticles to understand the ligand acquisition by a lipid bilayer or vesicle. Our group’s previously developed DPD modelling methods are used in the current studies. We evaluate the interactions between the gold nanoparticle and bilayer/vesicles that initiate the ligand acquisition.

Nonequilibrium Statistical Mechanics
We know the governing equations for atoms, though we can’t necessarily keep track of every atoms or describe how they assemble into molecules, aggregates, and even larger structures of a cell. While Feynman recognized that there was a lot of room—that is combinatoric complexity—at the atomic scale, we see that the combinatorics increase quickly at the middle scales where a myriad of heterostructures (of sub micron size) have been assembled and aggregated. We are developing approaches to scale the equations of motion with dynamical consistency so that we can indeed character nonequilibrium chemical dynamics in heterogeneous and complex environments. This includes our aim to characetrize sustainable nanoparticles that have emergent function at the middle scales, and characterizable function leading to non-toxic exposure in the environment. It also includes polymer-networked nanoparticle arrays which exhibit computing functions at the middle scales.
SPIRAL Project
Details in preparation…
- Nikhil Thota
- Maitreyee Sharma
Transition State Theory
The rates of chemical reactions (or any activated process) are by definition determined by the flux of reactants (or initial states) that end up as products (or final states). Through the last hundred years of studies on reaction rate theory, it has become clear that this can be equated to the flux through any surface that divides reactants from products as long as only those trajectories that end up as products are included in the flux. Transition state theory (TST) ignores this last clause. That is, it overestimates the rate if any of the trajectories recross the dividing surface. However, its advantage is that it replaces a dynamical calculation with a geometric one. Through a variational principle or perturbation theory, however, one can construct non-recrossing dividing surfaces that lead to exact rates. [Chem. Phys. 370, 270-276 (2010)] These approaches are limited by the nature of the search space of surfaces and the reference dividing surface, respectively. Nevertheless, we can determine such dividing surfaces geometrically using non-perturbative approaches. [J. Chem. Phys. 155, 210901 (2021)] Moreover, we can also address reactions under conditions far from equilibrium in so-called complex environments.
- Jörg Main, Collaborator
- Johannes Reiff, Collaborator
Open Chemistry Collaborative in Diversity Equity (OXIDE)
In the OXIDE project, we are working with academic chemistry department heads and chairs across the US to create and implement policies and procedures for advancing diversity equity and inclusion. We have published our findings on effective practices and conduct yearly surveys of the gender and URPOC demographics of research-active chemistry departments. We also run biennial National Diversity Equity Workshops (NDEWs) convening department chairs, DEI champions, social scientists, and other subject matter experts.
- Dontarie Stallings, Collaborator
- Vartika C. Saman