
It is well appreciated that experience leaves a lasting trace in the brain by modifying the efficacy of synaptic transmission between neurons. Such synaptic modifications occur throughout childhood to allow proper maturation of the brain in tune with the environment. And the same mechanisms ensure acquisition and storage of memory throughout life. Therefore, a fundamental question in neuroscience is to understand how experience alters synapses in the brain. The Lee lab approaches this question using multidisciplinary approaches combining electrophysiology, molecular/biochemistry, optogenetics, and imaging methods to study the changes that occur at synapses when a sensory modality is lost. Furthermore, we have been applying the current knowledge on the biological basis of synaptic plasticity to devise ways to treat memory disorders and restore brain function following sensory loss.
How is sensory loss compensated in the brain? – Cellular mechanisms of cross-modal plasticity
Sensory information arising from each sense organ is initially processed by well-defined areas of the brain, which are called primary sensory cortices. Traditionally, it was thought that each sensory modality was processed independently, however, there is emerging evidence that sensory integration occurs across different senses even at the early stages, such as the primary sensory cortices. The functionality of the sensory cortices can be profoundly changed by sensory experience, especially during early postnatal development. For example, loss of vision through one eye early in life causes “cortical blindness”, such that even if the optics of the eye is restored later in life, the individual is functionally blind through that eye. In essence, the brain loses its ability to “see” even though the eye is functional. It is also known that a complete loss of vision can lead to compensatory changes in the remaining sensory modalities to allow the blind individual to better utilize the remaining senses. However, the cellular and molecular correlates of cross-modal plasticity are poorly understood. The predominant view in the field is that cross-modal sensory compensation takes a long time to develop or that it requires extensive lesions early in development. However, contrary to this view, we found that within days after losing vision cross-modal plasticity can be detected in primary sensory cortices as functional changes in excitatory synaptic transmission (Goel et al. 2006 Nature Neuroscience). Based on these results we propose that the synaptic plasticity observed in the visual cortex, as well as cross-modally in other sensory cortices, may be a cellular correlate of cross-modal plasticity observed in blind individuals. Interestingly, while the cross-modal synaptic plasticity depends on sensory input to the remaining modalities, we did not find evidence of increased sensory input (He et al. 2012 Journal of Neuroscience). Furthermore, we found that the feed-forward thalamocortical synapses potentiate cross-modally after losing a sensory modality in adults without changes in the respective primary sensory cortex (Petrus et al. 2014 Neuron). This suggests that cross-modal sensory deprivation is more effective at recruiting thalamocortical plasticity in the adult cortex. Consistent with this idea, we recently demonstrated that deafening adults for a short duration can recover rapid ocular dominance plasticity in the primary visual cortex mainly via restoration of thalamocortical LTP (Rodriguez et al. 2018 Cell Reports). Our results suggest that cross-modal plasticity may be used as a tool to recover plasticity in the adult brain.
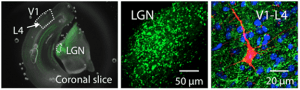
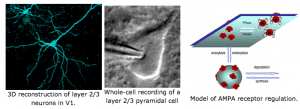
Molecular mechanisms of sensory cortex plasticity
At a molecular level, we found that sensory experience alters synaptic AMPA receptor function. AMPA receptors are activated by glutamate, which is the major excitatory neurotransmitter in the brain. Therefore, any change in AMPA receptor function will alter information transfer between neurons, and result in changes in the functionality of the neural circuits. We specifically found that loss of vision up-regulates excitatory synaptic transmission in the visual cortex, which correlates with the appearance of Ca2+ -permeable AMPA receptors at synapses and phosphorylation of AMPA receptor subunit GluR1 (or GluA1) at S845. One role of GluR1-S845 phosphorylation is to stabilize Ca2+-permeable AMPA receptors at perisynaptic sites by preventing their degradation (He et al. 2009 Proc. Natl. Acad. Sci-Fi. USA). The increase in GluR1-S845 phosphorylation is interesting, because we know from our collaboration with Dr. Alfredo Kirkwood’s lab at Johns Hopkins that it selectively enhances activity-dependent strengthening of visual cortex synapses (Seol et al. 2007 Neuron). While AMPA receptor phosphorylation seems critical for homeostatic up-regulation of synaptic strength, we have evidence that homeostatic weakening of synapses may not be a simple reversal of this process. In collaboration with Dr. Paul Worley’s group at Johns Hopkins, we found that an immediate early gene product Arc (activity regulated cytoskeleton-associated protein) and Homer1a are critically involved. Both Arc and Homer1a expression can readout salient stimulation suggesting that they may be involved in homeostatic reduction of excitatory synaptic transmission triggered by enhanced sensory input. Consistent with this, we found that indeed visual experience-dependent homeostatic synaptic plasticity is absent in Arc knockout mice (Gao et al. 2010 Journal of Neuroscience) and mice lacking Homer1a signaling (Chokshi et al., 2019 Neuron).
In parallel to excitatory synapses, we found that sensory experience regulates inhibitory synapses in a distinct manner. Visual experience is critical for the developmental maturation of inhibitory synapse number (Kreczko et al. 2009 Neural Plasticity) and synaptic GABA-A receptor number and function (Gao et al. 2014 Journal of Neuroscience). In addition, gap junctions play a role in regulating inhibitory synaptic transmission (Postma et al. 2011 Proc. Natl. Acad. Sci. USA). In collaboration with Dr. Alfredo Kirkwood, we found that LTD of inhibitory synapse utilizes presynaptic regulation of release via retrograde endocannabinoid signaling (Jiang et al. 2010 Neuron). In contrast, homeostatic regulation of inhibitory synapses occurs via postsynaptic regulation of GABA-A receptors, which manifests as potentiation following recovery of vision in juveniles (Gao et al. 2014 Journal of Neuroscience). Distinct from juveniles, in adults visual experience specifically regulates action potential independent inhibitory synaptic release, which we determined is critical for preserving information coding in a mature cortical circuit (Gao et al. 2017 Phil. Trans. Royal Soc. B).
Interestingly, the homeostatic synaptic changes induced by visual deprivation occurred throughout development into adulthood in the superficial layers of the visual cortex (Goel and Lee 2007 Journal of Neuroscience). The significance of this finding is that it suggests that synapses in the adult cortex retain the ability to undergo functional alterations with relatively brief durations of change in visual experience. While the homeostatic synaptic changes persisted in the adult visual cortex, we found evidence that the mechanisms underlying adult and juvenile plasticity may be different. In particular, our data suggest that adult plasticity may be more restricted than juveniles, and also highlights the possibility that there may be specific inputs that retain the ability to support experience-dependent synaptic plasticity in adults. In particular, we found evidence that intracortical synapses are much more plastic than synapses serving the feedforward pathway that brings in sensory information to the cortex (Petrus et al. 2015 Journal of Neuroscience). At a molecular level, sensory experience dependent homeostatic synaptic plasticity are dependent on NMDA receptors (Rodriguez et al., 2019 Journal of Neuroscience ) and metabotropic glutamate receptor 5 (mGluR5) signaling (Chokshi et al., 2019 Neuron). We hypothesize that the plasticity is due to a metaplasticity mechanism termed sliding threshold, which allows the cortical neurons to adapt their feature selectivity in tune with the sensory environment and the internal state (Lee & Kirkwood, 2019 Frontiers in Cellular Neuroscience).
Efforts to develop effective therapies for the treatment of Alzheimer’s disease
We are also applying our knowledge of activity-dependent synaptic plasticity to develop treatments for Alzheimer’s disease. Alzheimer’s disease (AD) is the most prevalent form of senile dementia affecting about 1 in 8 seniors above the age of 65. Currently, there is no effective treatment to stop the progression of the disease. It is widely believed that AD is initiated as a synaptic dysfunction, which correlates with the loss of memory function in the early stages of the disease. A current working hypothesis in the field is that over-production of amyloid-beta (Aß) peptide initiates the pathogenesis of AD. Therefore, inhibition of enzymes that produce Aß have emerged as promising targets to treat AD. A transmembrane aspartic protease, called BACE1 (beta-site APP cleavage enzyme 1), has been identified as one of the key enzymes involved in Aß production. The amount and activity of BACE1 is elevated in sporadic AD brains, which is the majority of AD cases, further suggesting that effective methods to reduce its activity may be beneficial to a large population of AD patients. Several laboratories, including our collaborator Dr. Philip Wong’s at Johns Hopkins Medical School, generated BACE1 knockout mice to determine the functional consequences of chronic inhibition of BACE1 activity. As predicted, BACE1 knockouts do not produce A ß. However, we found that BACE1 knockouts display specific presynaptic deficits, which correlated with behavioral dysfunctions (Laird et al. 2005 Journal of Neuroscience). Our work suggests that a complete ablation of BACE1 activity may be deleterious for normal synaptic function and will produce abnormal behavior. In order to develop BACE1 inhibitors as effective treatment options for AD, it is critical to determine ways to circumvent the unwanted functional deficits seen in the BACE1 knockouts. We found that BACE1 knockouts fail to undergo activity-dependent strengthening of presynaptic release at the mossy fiber synapses in the CA3 region of the hippocampus, which was specifically due to impaired presynaptic Ca2+ signaling (Wang et al. 2008 Journal of Neuroscience). Incidentally, this set of synapses express the highest level of BACE1 protein in the brain. Our results indicate a previously unsuspected role of BACE1 in the regulation of presynaptic release. Recently, we found that activation of alpha7-nicotinic acetylcholine receptors (alpha7-nAchRs) can elevate presynaptic release and “rescue” the BACE1 knockout phenotype (Wang et al. 2010 Journal of Neuroscience). Our results suggest that presynaptic dysfunctions associated with BACE1 inhibition can be circumvented via pharmacological means, which we expect would benefit the development of AD therapies based on BACE1 inhibition. In parallel, we are examining other potential drugs and molecular interventions that can alleviate synaptic dysfunctions in a mouse model of Alzheimer’s disease (Megill et al. 2013 Journal of Neuroscience), and have identified metaplasticity dysfunction as a potential culprit that may be fundamental to synaptic plasticity deficits (Megill et al. 2015 Journal of Neuroscience).
